INTRODUCTION
Plexiform angiomyxoid myofibroblastic tumour was first reported and described in 2007 by Takahashi et al.1 They reported two cases of a gastric tumour with similar morphology that they called “plexiform angiomyxoid myofibroblastic tumour.” In 2009, Miettinen et al.2 reported and described twelve additional cases of plexiform angiomyxoid myofibroblastic tumours located in the gastric antrum. These authors were also the first to use the diagnostic term “plexiform fibromyxoma” instead of “plexiform angiomyxoid myofibroblastic tumour.” In 2010, plexiform fibromyxoma was included in the “Mesenchymal tumours of the stomach” category of the 4th edition of the WHO Classification of Tumours of the Digestive System.3–5 In 2019, plexiform fibromyxoma was included in the “Adipose tissue and (myo)fibroblastic tumours” category of the 5th edition of the WHO Classification of Tumours of the Digestive System.6 Plexiform angiomyxoid myofibroblastic tumour is a term accepted by the 5th WHO classification.6 Since the first cases reported by Takahashi et al.,1 many further cases have been reported in the literature. To date, 138 cases of plexiform fibromyxoma have been reported.7 Males and females are equally affected.3 The majority of the cases have been found in adults with a median age of 40 to 50 years, but there are pediatric cases reported.3,6,8 The most common site for plexiform fibromyxoma is the gastric antrum.3,6,8 However, three cases have been reported in the oesophagus,9 one case in the gallbladder,9,10 one case in the mediastinum,7 and three cases in the duodenum.11–13 Zhang et al.14 reported a case of plexiform fibromyxoma located in the upper jejunum, 100 cm distal to the duodenal papilla. This was in a 31 year-old woman with repeated hematochezia and anemia. No other plexiform fibromyxoma located in the distal small bowel has been reported in the literature.
CASE PRESENTATION
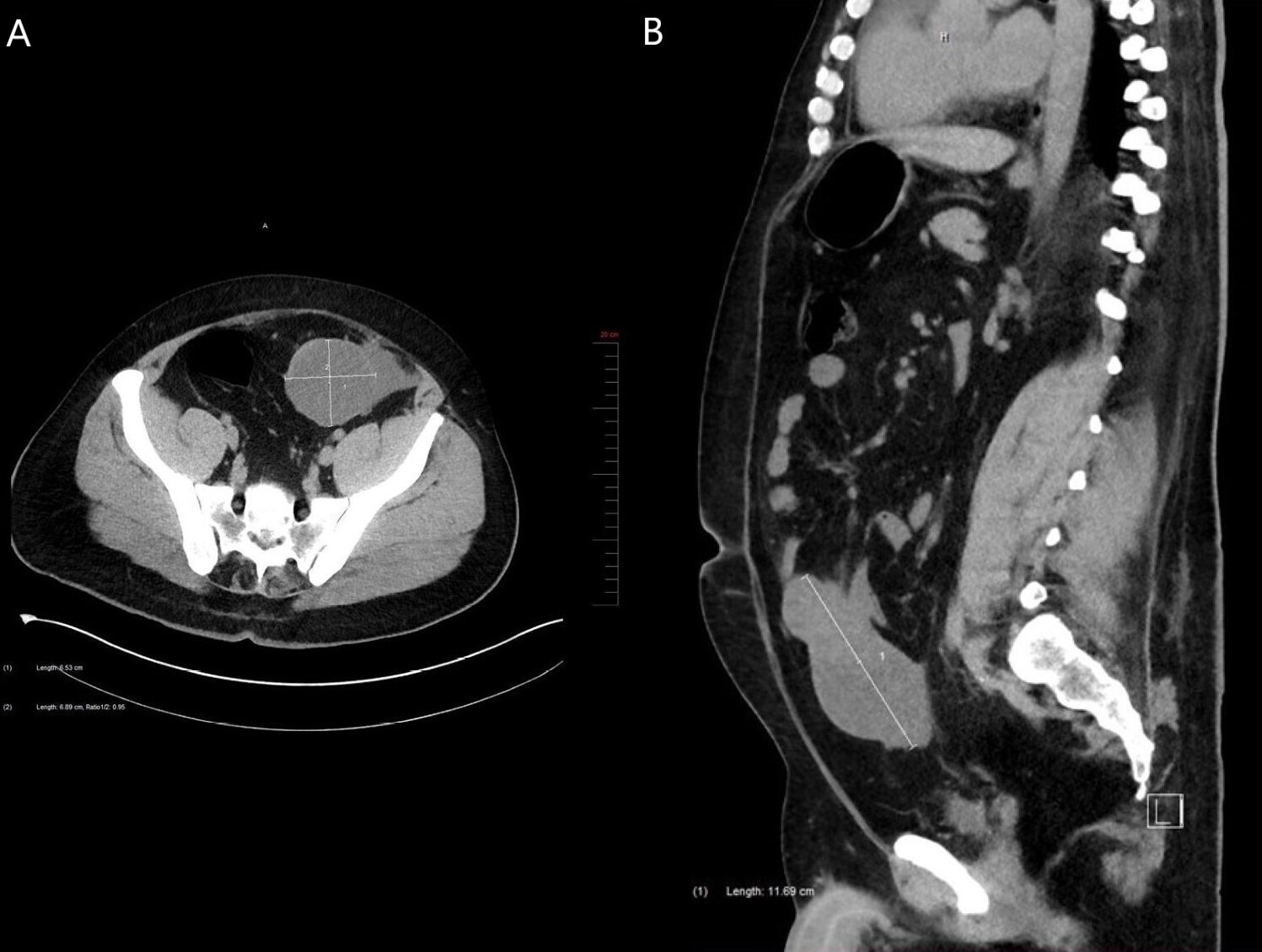
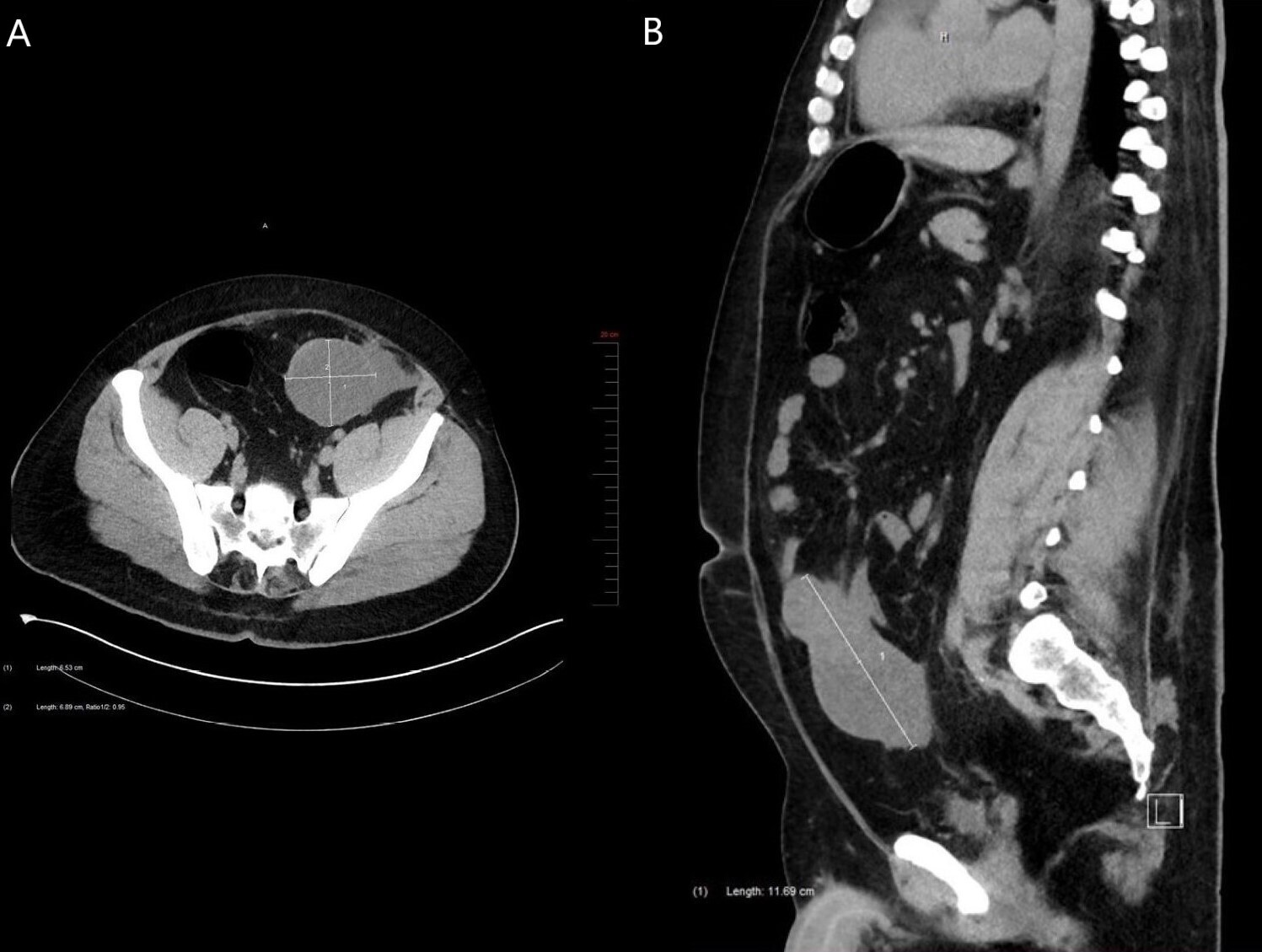
A 36-year-old man presented with vague abdominal pain and iron-deficiency anemia. His medical history included sleep apnea, hypothyroidism, gastroesophageal reflux disease, Steinert myotonic dystrophy syndrome and a non-secreting left adrenal adenoma measuring 2.2cm. His iron-deficiency anemia was investigated by colonoscopy, which was normal. One year later, the patient went to the emergency room for abdominal pain. A CT scan revealed chronic calculous cholecystitis and a tumour. This tumour was a mixed solid and cystic polylobulated mass measuring 11.7cm in its greatest dimension and located in the intraperitoneal fat at the lower portion of the greater omentum along the midline (Figures 1A and 1B). The tumour was adjacent to the small bowel and the colon, and there was an absence of lymphadenopathy. Gastrointestinal stromal tumour (GIST), desmoid fibromatosis and small intestinal lymphoma were clinically suspected. Subsequently, the patient underwent diagnostic laparoscopy which demonstrated a tumour located in the small bowel. Cholecystectomy and distal small bowel resection without colon resection were performed in the same procedure. The patient had no complications.
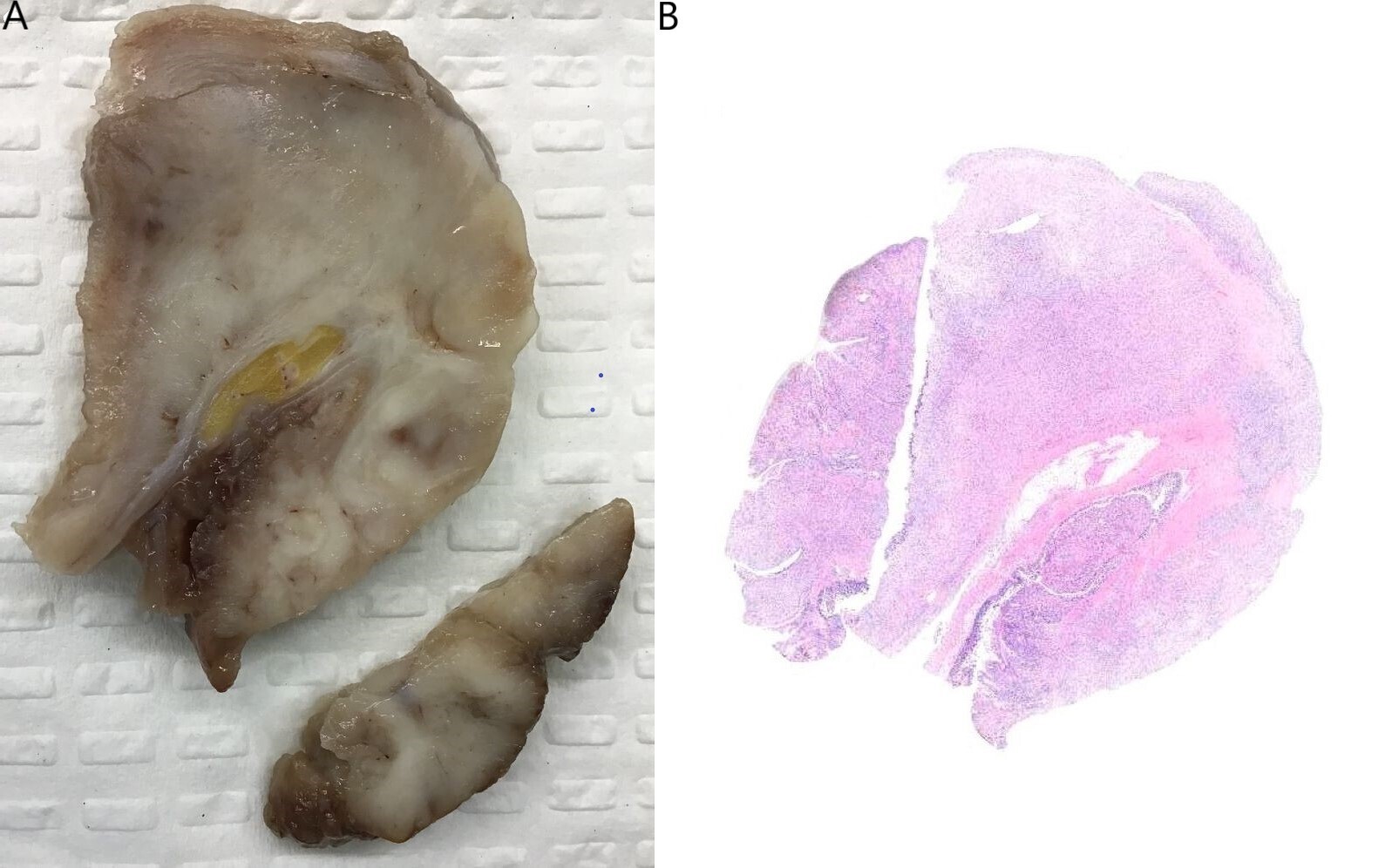
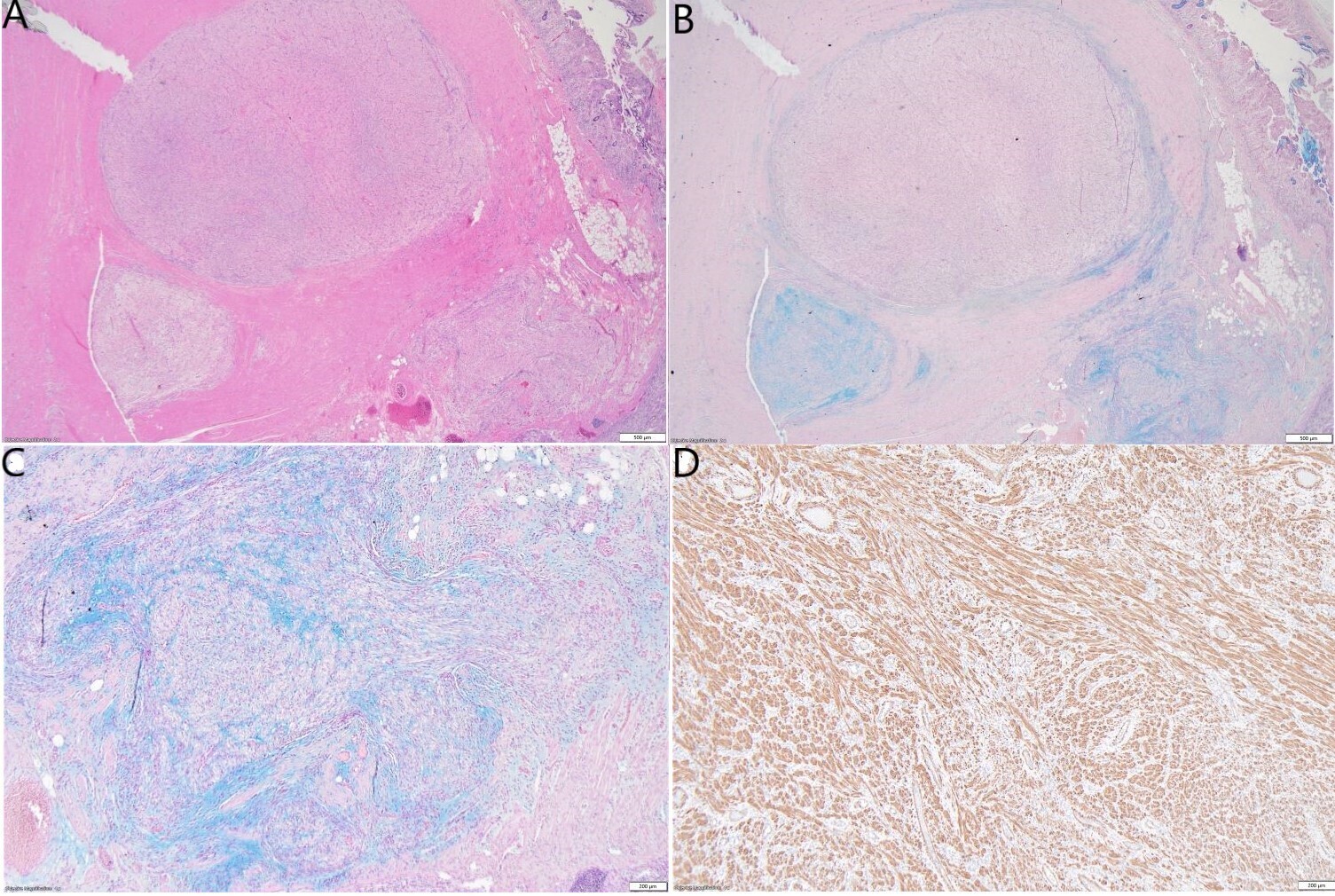
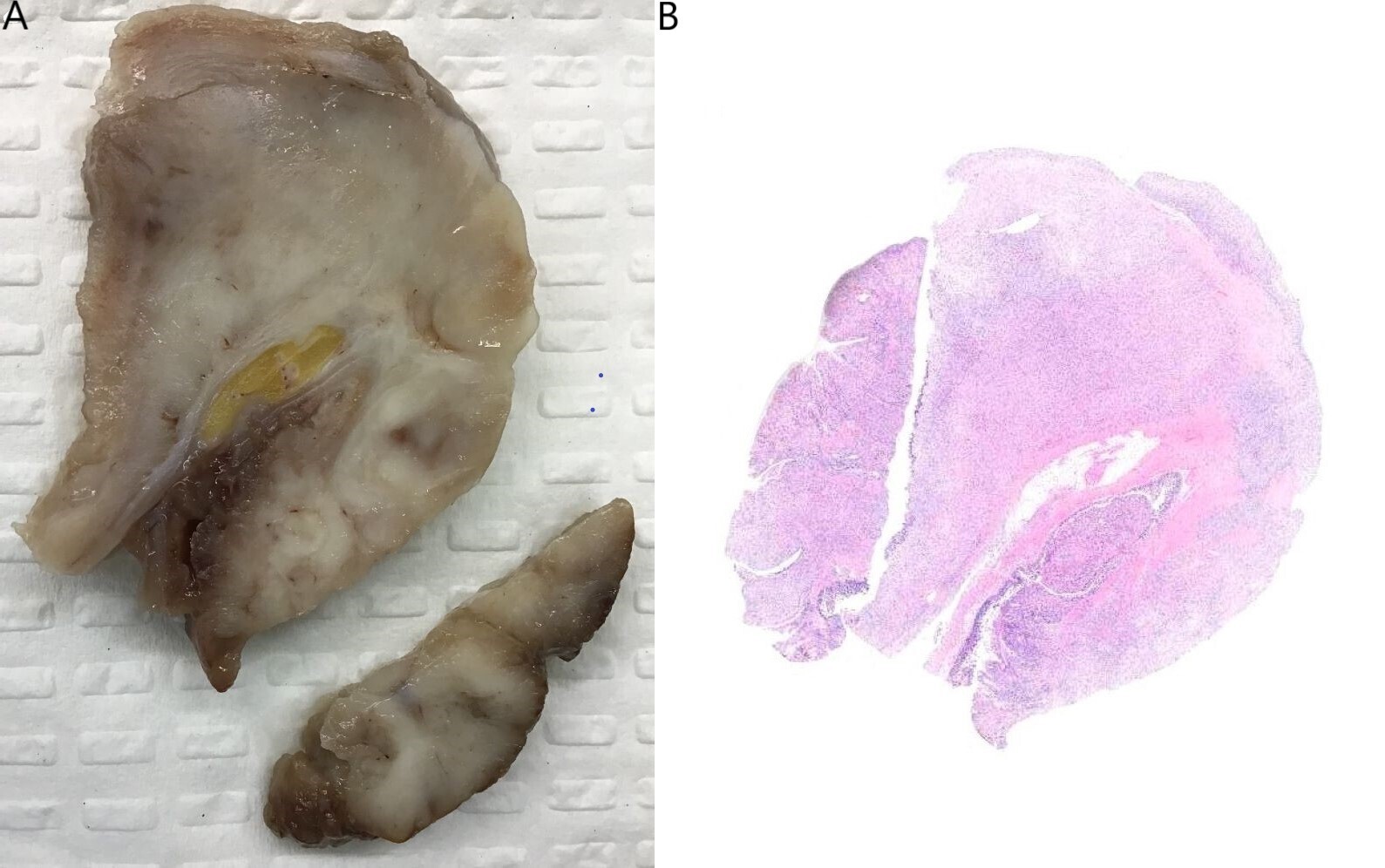
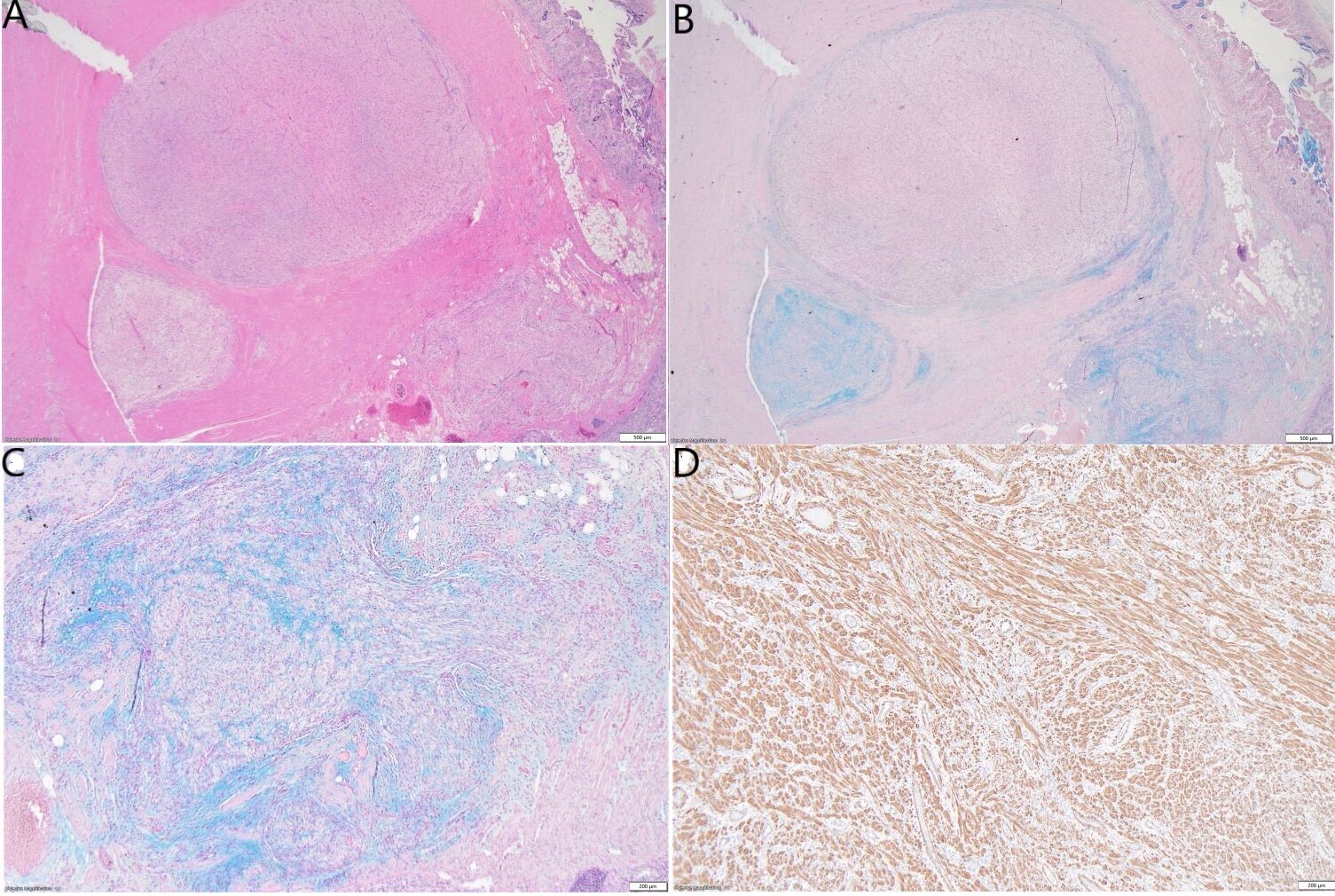
Grossly, the small bowel resection demonstrated a 10.6cm solid white tumour with a fascicular appearance infiltrating the entire thickness of the small bowel wall. The mass protruded into the intestinal lumen and occupied 90% of the intestinal circumference. The intestinal wall had also a focal mucosal invagination encompassed by the tumour (Figures 2A and 2B). Histologically, the tumour demonstrated a proliferation of bland spindle cells with a multinodular and plexiform architecture (Figures 3A and 3B) centered in the muscularis propria and involving the lamina propria, submucosa and subserosa. The mucosa above the lesion was focally ulcerated, with granulation tissue.
The stroma in the nodules was fibromyxoid (Figures 3B and 3C) with a proliferation of arborizing capillaries. Neither cytologic atypia, necrosis, nor increased mitoses were noted. The adipose tissue and soft tissues adjacent to the tumour showed no cytological atypia and no significant histological alterations. Immunohistochemical studies demonstrated that the spindle cells were immunoreactive with smooth muscle actin (SMA), caldesmon and calponin (Figure 3D). They were not immunoreactive with desmin, CD34, CD117, DOG1, B-catenin, Myoglobin, SOX10, S100, Melan A, HMB45, STAT6, ALK1, CDX2, EMA, CK AE1/AE3, MUC2, MUC6, ERG, CD31 or MDM2. Ki-67 labelling was low (<3%). SDH-B was retained. Special stains with Alcian Blue and PAS showed focal mucinous deposition (Figure 3B and Figure 3C).
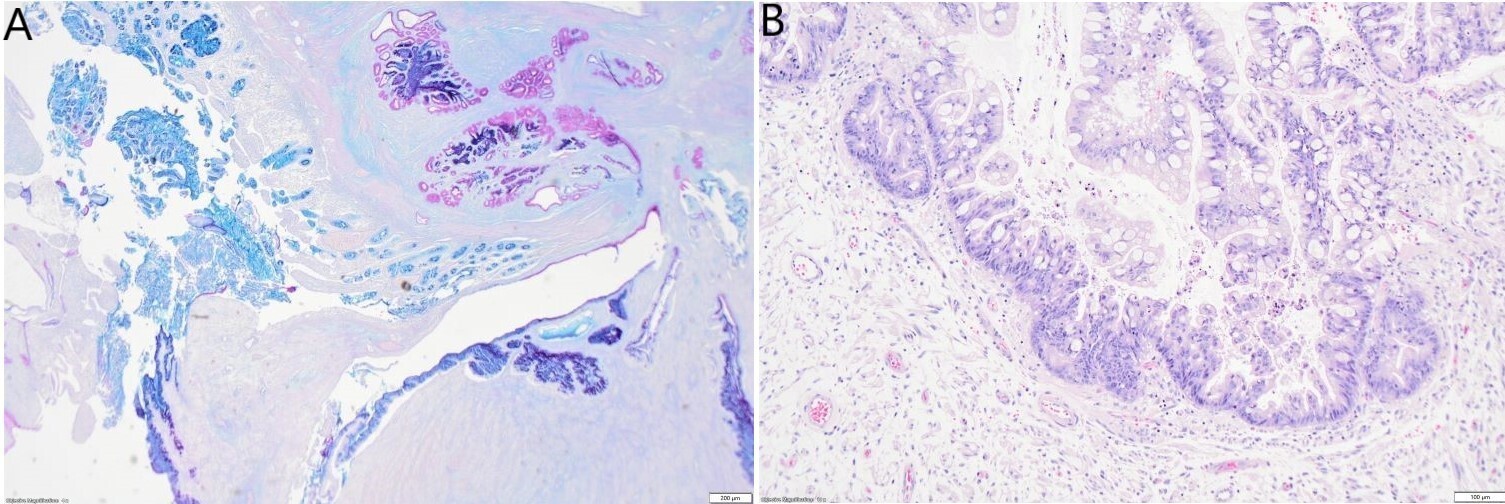
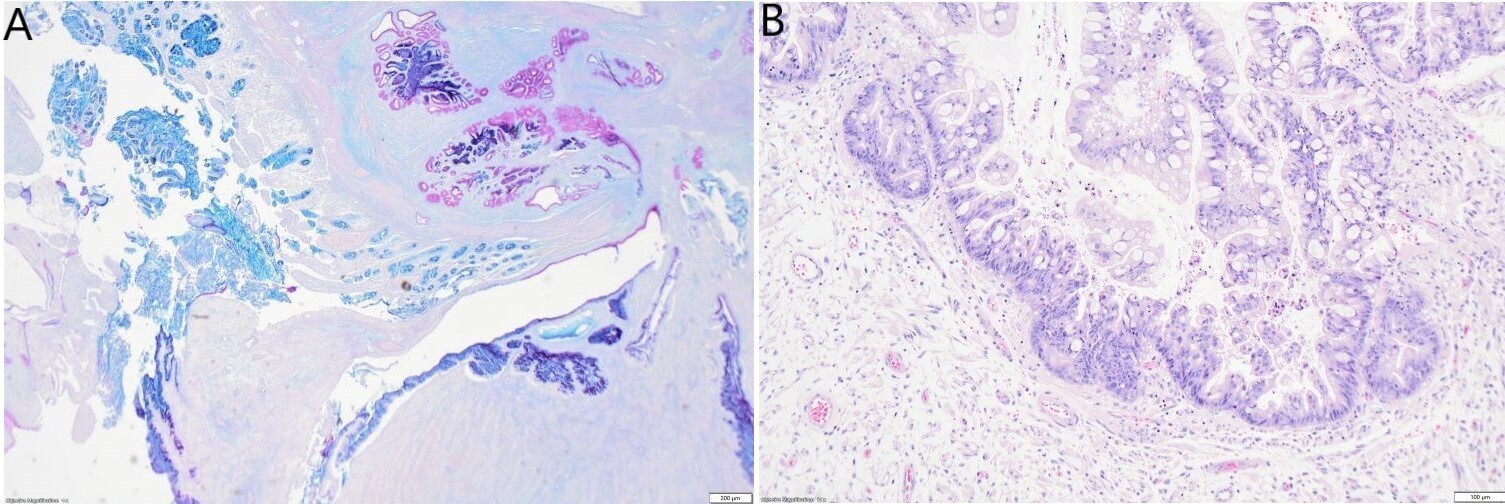
In some slides, the small intestinal wall also demonstrated a mucosal invagination encompassed by the tumour. This mucosal invagination formed a pseudo-cavity with a mixed epithelium composed of intestinal epithelium which was positive for MUC2 and MUC6, and metaplastic or heterotopic gastric antral epithelium which was positive for EMA, MUC5AC and PAS (Figure 4A). The epithelium in the mucosal invagination showed foci of high-grade epithelial dysplasia without evidence of infiltrating malignancy (Figure 4B).
Eleven lymph nodes were identified and none had any metastasis. The surgical margins were free of tumour.
Additional molecular testing, including MDM2 FISH and a sarcoma-targeted RNA sequencing panel, was performed on the tumour. The analyses revealed no evidence of MDM2 gene amplification and no other significant molecular alterations. Neither MALAT1–GLI1 fusion nor sequence variants were detected.
Morphologic, immunohistochemical and molecular features were consistent with a diagnosis of plexiform fibromyxoma located in the distal ileum and encompassing a probable Meckel diverticulum with foci of high-grade epithelial dysplasia. To this day, two years after the surgery, the patient has not experienced a recurrence of his tumour.
DISCUSSION
Plexiform fibromyxoma is a rare mesenchymal tumour with a nonspecific clinical presentation.3 To date, 138 cases of plexiform fibromyxoma have been reported.7 The most common site for this tumour is the gastric antrum.3,6,8 However, three cases have been reported in the oesophagus,9 one case in the gallbladder,9,10 one case in the mediastinum,7 three cases in the duodenum,11–13 and one case in the upper jejunum 100 cm distal to the duodenal papilla.14 No cases in the distal small bowel or associated with a Meckel diverticulum have been described. The distal small bowel plexiform fibromyxoma described in this case report was encompassed by a probable Meckel diverticulum. The presence of both the plexiform fibromyxoma and the Meckel diverticulum at the same time could be a result of chance, with no association between them. Since the majority of plexiform fibromyxomas are located in the stomach,3,6,8 it can be hypothesized that the Meckel diverticulum with gastric heterotopia may have been a risk factor for the development of the tumour.
On CT scan or magnetic resonance imaging (MRI), plexiform fibromyxoma presents as a solid, cystic, or solid and cystic mass with well-defined borders.3 CT scan may also show mild enhancement of the solid portion during the arterial phase and progressive enhancement during the venous and delayed phases.3 On macroscopic examination, plexiform fibromyxoma demonstrates a multinodular mass with myxoid or gelatinous appearance.3,6,8 There can be foci of hemorrhage.3,6,8 The mass is centered in the muscularis propria but it can extend in the submucosa and in the mucosa, causing ulceration.3,6,8 The mass can also project on the serosa.6,8
Histologically, plexiform fibromyxoma is characterized by a proliferation of bland spindle to ovoid cells with a plexiform and multinodular architecture.3,6,8 The stroma is myxoid, fibromyxoid or collagenous with arborizing proliferation of thin-wall capillaries.3,6,8 There are few mitoses, no atypia, and no necrosis.3,6,8 Mast cell infiltration may be present.8
Immunohistochemically, the tumour cells are positive for smooth muscle actin (SMA) and vimentin.3,6,8 They may be positive for CD10, desmin, caldesmon and calponin. However, they are not positive for DOG1, CD117 (C-Kit), EMA, ALK, S100, CD34, and beta-catenin. The Ki-67 index is low, and Alcian Blue stain may demonstrate the myxoid background.3
Molecular features of plexiform fibromyxoma have been recently described in the literature, although they are not specific to this tumour. Approximately 18% of cases may present with MALAT1–GLI1 gene fusions.3,6 Other molecular alterations that can occur in some cases of plexiform fibromyxoma include GLI1 polysomy or PTCH1 inactivation through gene or chromosomal deletion.3,6 Other lesions with spindle cell proliferation and/or myxoid stroma must be ruled out before establishing a diagnosis of plexiform fibromyxoma. Histologic mimics include GISTs (spindle cell GISTs, myxoid GISTs and succinate-dehydrogenase-deficient GISTs), smooth muscle tumours (leiomyomas and leiomyosarcomas), schwannomas, desmoid fibromatosis, solitary fibrous tumours, inflammatory myofibroblastic tumours and inflammatory fibroid polyps.
Gastrointestinal GISTs exhibit a proliferation of spindle cells, epithelioid cells, or a mix of spindle and epithelioid cells.15 Collagen globules (skenoid fibers) and prominent paranuclear vacuoles can be present. The stroma can be myxoid or hyalinized. SDH-deficient GISTs usually arise in the stomach and demonstrate a multinodular growth pattern of epithelioid cells with a loss of cytoplasmic SDHB staining by immunohistochemistry.15 GISTs are positive for CD117 and DOG1 (95% of cases), CD34 (70% of cases) and caldesmon (70% of the cases). 30-40% express SMA; rare GISTs are positive for desmin, keratins or S100.6,15,16
Gastrointestinal leiomyomas exhibit fascicles of bland spindle cells with blunt-ended nuclei, eosinophilic cytoplasm, low mitotic activity, and no necrosis.3,6 Gastrointestinal leiomyosarcomas have cellular fascicles of spindle cells with atypia, high mitotic activity, and with or without necrosis.3,6 Leiomyomas and leiomyosarcomas can have a myxoid background and both are positive for desmin, SMA, caldesmon and calponin. They are negative for DOG1, CD117, CD34 and S100.3,6
Gastrointestinal schwannomas are well-circumscribed unencapsulated mural nodule with peritumoural lymphoid cuffs.3,6 The tumour cells are spindle and have a microtrabecular architecture. They are strongly positive for S100 but are negative for CD34, CD117, DOG1, SMA and desmin.3,6
Desmoid fibromatosis arises in the mesentery of small bowel as an infiltrating mass with long sweeping fascicles of uniform and bland spindle or stellate myofibroblasts.6,8 The stroma can be collagenous or myxoid. 80% of desmoid fibromatosis have nuclear B-catenin staining and SMA is normally positive.6,8
Solitary fibrous tumours may arise at any anatomical location as a lesion with poorly defined fascicles (“patternless pattern”) of uniform spindle or ovoid fibroblastic cells. The vascular network is prominent and displays staghorn vessels with perivascular hyalinization.6,8 The stroma can be fibrous or myxoid. 98% of solitary fibrous tumours have strong nuclear STAT6 positivity and 90-95% have strong CD34 positivity.6,8
Gastrointestinal inflammatory myofibroblastic tumours have spindle or stellate myofibroblastic cells in a loose fascicular pattern. The stroma is myxoid or collagenous with a mixed inflammatory infiltrate. Mitotic activity is low and there is no necrosis. The tumour cells are positive for SMA, 50% are positive for desmin and 60% are positive for ALK (diffuse cytoplasmic staining). CD117, DOG1, CD34, S100 and SOX10 are negative.6,8
Inflammatory fibroid polyps are benign hypocellular fibroblastic neoplasm with bland spindle and stellate cells. The stroma is edematous, myxoid or collagenous with a prominent mixed inflammatory infiltrate rich in eosinophils and lymphocytes. There is also a proliferation of blood vessels with concentric fibrosis (onion skin fibrosis). CD34 is positive. CD117, DOG1, Desmin, S100, SOX10 and keratins are negative.3,6
CONCLUSION
Here, we report the first case of plexiform fibromyxoma located in the distal ileum encompassing probably a Meckel diverticulum with foci of high-grade epithelial dysplasia without invasive carcinoma. It is important for surgical pathologists to be aware that this entity can be found in the distal small bowel. It should be included in the differential diagnosis of spindle cell proliferation and/or myxoid lesions located in the distal small bowel.